Le sildénafil présent dans Kamagra exerce une inhibition réversible de la PDE5, modulant la cascade GMPc et favorisant une vasodilatation localisée. L’absorption digestive varie selon la forme utilisée, comprimés classiques ou gels oraux. La distribution tissulaire est large et la liaison protéique élevée, avoisinant 96 %. La métabolisation hépatique génère un métabolite actif contribuant à l’effet pharmacologique global. La demi-vie reste courte, avec disparition plasmatique en quelques heures. Les interactions significatives concernent surtout les nitrés organiques et inhibiteurs puissants du CYP3A4. Dans les publications techniques, kamagra en ligne est souvent cité dans le cadre d’analyses comparatives portant sur les différences de formulations et de cinétique d’absorption.
No job name
Highly Selective Hydrolytic Kinetic Resolution of Terminal Epoxides Catalyzed by Chiral (salen)CoIII Complexes. Practical Synthesis of Enantioenriched Terminal Epoxides and 1,2-Diols
Scott E. Schaus, Bridget D. Brandes, Jay F. Larrow, Makoto Tokunaga,
Karl B. Hansen, Alexandra E. Gould, Michael E. Furrow, and Eric N. Jacobsen*
Department of Chemistry and Chemical Biology, HarVard UniVersity,
Received July 31, 2001. Revised Manuscript Received October 23, 2001
Abstract: The hydrolytic kinetic resolution (HKR) of terminal epoxides catalyzed by chiral (salen)CoIII complex 1‚OAc affords both recovered unreacted epoxide and 1,2-diol product in highly enantioenriched form. As such, the HKR provides general access to useful, highly enantioenriched chiral building blocks that are otherwise difficult to access, from inexpensive racemic materials. The reaction has several appealing features from a practical standpoint, including the use of H2O as a reactant and low loadings (0.2-2.0 mol %) of a recyclable, commercially available catalyst. In addition, the HKR displays extraordinary scope, as a wide assortment of sterically and electronically varied epoxides can be resolved to g99% ee. The corresponding 1,2-diols were produced in good-to-high enantiomeric excess using 0.45 equiv of H2O. Useful and general protocols are provided for the isolation of highly enantioenriched epoxides and diols, as well as for catalyst recovery and recycling. Selectivity factors (krel) were determined for the HKR reactions by measuring the product ee at ca. 20% conversion. In nearly all cases, krel values for the HKR exceed 50, and in several cases are well in excess of 200. Introduction
Since those epoxides that are produced naturally are typically
complex compounds available only in limited amounts, Nature’s
The importance of epoxides in organic synthesis arises partly
chiral pool has not proven to be a useful direct source of
from the occurrence of the strained three-membered ring unit
optically active epoxides for use in organic synthesis. Instead,
in a number of interesting natural products1 but more so because
enantioenriched epoxides have been accessed indirectly from
the ring opening of epoxides allows straightforward elaboration
the chiral pool via multistep procedures.3 These, however, tend
to useful new functionality, often with generation of new
to be inherently inefficient, and the range of epoxides available
carbon-carbon bonds. Indeed, reactions of epoxides with
by this approach is also quite limited. As a consequence, the
nucleophiles, Lewis acids, radicals, reducing agents, oxidizing
preparation of enantioenriched epoxides has long stood as a most
agents, acids, and bases have all been well documented and
significant target for asymmetric synthesis. In particular, the
utilized in synthesis.2 Further, the stereospecific manner in which
identification of catalytic asymmetric olefin oxidation methods
epoxides generally react renders these compounds attractive
has been an area of active research for several decades, and the
chiral building blocks for asymmetric synthesis.
advances made in this field have increased greatly the numberof enantiomerically enriched epoxides available for use in
(1) Some, among many, notable examples: (a) Fumagillin: Tarbell, D. S.;
Carman, R. M.; Chapman, D. D.; Cremer, S. E.; Cross, A. D.; Huffman,K. R.; Kuntsmann, M.; McCorkindale, N. J.; McNally, J. G.; Rosowsky,
Among available methods for the preparation of enantio-
A.; Varino, F. H. L.; West, R. L. J. Am. Chem. Soc. 1961, 83, 3096. (b)
enriched epoxides, the Sharpless epoxidation reaction has
Ovalicin: Sigg, H. P.; Weber, H. P. HelV. Chim. Acta 1968, 51, 1395. (c) Coriolin: Takeuchi, T.; Iinuma, H.; Iwanaga, J.; Takahashi, S.; Takita, T.;
arguably had the most profound impact of any asymmetric
Umezawa, H. J. Antibiot. 1969, 22, 215. (d) Disparlure: Bierl, B. A.; Beroza, M.; Collier, C. W. Science 1970, 170, 87. (e) Triptolide: Kupchan, S. M.; Court, W. A.; Dailey, R. G.; Gilmore, C. J.; Bryan, R. F. J. Am.
(2) For reviews and lead references, see: (a) Winstein, S.; Henderson, R. B. Chem. Soc. 1972, 94, 7194. (f) Periplanone B: Persoons, C. J.; Verwiel,
In Heterocyclic Compounds, Vol. 1; Elderfield, R. C., Ed.; Wiley: New
P. E. J.; Ritter, F. J.; Talman, E.; Nooijen, P. J.; Nooijen, W. J. Tetrahedron
York, 1950; Chapter 1. (b) Parker, R. E.; Isaacs, N. S. Chem. ReV. 1959, Lett. 1976, 17, 2055. (g) Neocarzinostatin chromophore: Edo, K.; Mizugaki, 59, 737. (c) Barto´k, M.; La´ng, K. L. Small Ring Heterocycles. In The
M.; Koide, Y.; Seto, H.; Furihata, K.; Otake, N.; Ishida, N. TetrahedronChemistry of Heterocyclic Compounds, Vol. 42, Part 3; Hassner, A., Ed.;
Lett. 1985, 26, 331. (h) Trapoxins: Itazaki, H.; Nagashima, K.; Sugita, K.;
Wiley: New York, 1985; Chapter 1. (d) Rao, A. S.; Paknikar, S. K.; Kirtane,
Yoshida, H.; Kawamura, Y.; Yasuda, Y.; Matsumoto, K.; Ishii, K.; Uotani,
J. G. Tetrahedron 1983, 39, 2323. (e) Smith, J. G. Synthesis 1984, 629.
N.; Nakai, H.; Terui, A.; Yoshimatsu, S. J. Antibiot. 1990, 43, 1524. (i)
(3) For examples, see: (a) Larcheveˆque, M.; Petit, Y. Tetrahedron Lett. 1987,
Epothilones: Bollag, D. M.; McQueney, P. A.; Zhu, J.; Hensens, O.;
28, 1993. (b) Larcheveˆque, M.; Henrot, S. Tetrahedron 1990, 46, 4277.
Koupal, L.; Liesch, J.; Goetz, M.; Lazarides, E.; Woods, C. M. Cancer
(c) de March, P.; Figueredo, M.; Font, J.; Monsalvatje, M. Synth. Commun.Res. 1995, 55, 2325. (j) FR901464: Nakajima, H.; Takase, S.; Terano, H.; 1995, 25, 331. (d) Adiyaman, M.; Khanapure, S. P.; Hwang, S. W.; Rokach,
Tanaka, H. J. Antibiot. 1997, 50, 96.
J. Tetrahedron Lett. 1995, 36, 7367. 10.1021/ja016737l CCC: $22.00 2002 American Chemical Society J. AM. CHEM. SOC. 9 VOL. 124, NO. 7, 2002 A R T I C L E S
catalytic reaction discovered thus far, providing general access
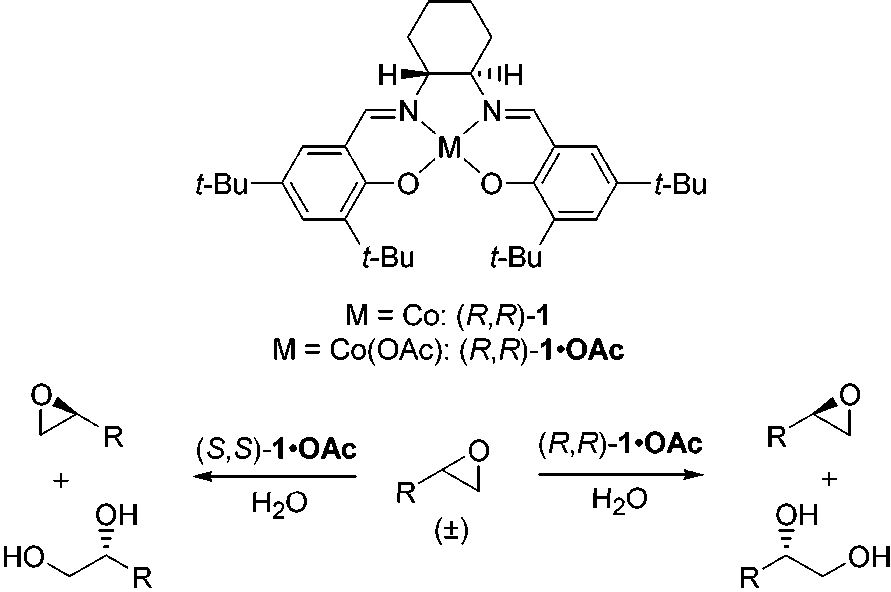
Scheme 1. Hydrolytic Kinetic Resolution (HKR) Reaction
to highly enantioenriched epoxyalcohols.4 More recently, theepoxidation of unfunctionalized conjugated olefins by chiral(salen)MnIII complexes has enabled the practical synthesis ofcertain classes of enantiomerically enriched epoxides.5 A highlycomplementary strategy for epoxidation of simple olefinsinvolving chiral dioxirane intermediates has expanded the rangeof chiral epoxides now accessible in enantioenriched form to asignificant extent.6 Indirect routes to enantiopure epoxidesinvolving asymmetric catalytic dihydroxylation or reductionreactions have also proven highly valuable in specific contexts.7
Despite these considerable advances in asymmetric catalytic
synthesis of epoxides, to date no general methods have beenidentified for the direct preparation of highly enantioenriched1-oxiranes, arguably the most valuable class of epoxides for
composition of unreacted substrate can be controlled by
organic synthesis.8 The utility of terminal epoxides as chiral
adjusting the degree of conversion, and virtually enantiopure
building blocks is perhaps best illustrated by the fact that the
material can be obtained at appropriately high conversions.14
few examples for which effective catalytic approaches exist have
This is an important consideration in the present case, since
found extensive use in asymmetric synthesis. In particular,
low-molecular weight terminal epoxides are typically liquids
glycidol and a number of its derivatives are available in
at room temperature and are not readily derivatized as salts,
enantiomerically enriched form using the Sharpless epoxidation
and therefore it is not a straightforward matter to upgrade their
technology9 or by enzymatic kinetic resolution methods,10 and
enantiomeric composition by crystallization. However, in the
these compounds have become widely used starting materials
absence of straightforward substrate racemization protocols,
for target-oriented synthesis.11 Epichlorohydrin has been ren-
kinetic resolutions have the significant disadvantage of a 50%
dered commercially available in bulk by microbial resolution
maximum yield of substrate recovery. With a specific interest
of (()-2,3-dichloro-1-propanol,12 and it, too, has found wide-
in devising a practical method for obtaining highly enantio-
enriched terminal epoxides, we deemed that the following
Pursuant to our own efforts directed toward the development
criteria must be met in order for a kinetic resolution approach
of catalysts for the enantioselective nucleophilic ring opening
of meso epoxides,13 we became interested in the possibility of
(1) The racemic epoxides must be inexpensive or easily
developing analogous methodology for the kinetic resolution
accessible from inexpensive commercial starting materials.
of 1,2-epoxides. One of the most attractive features of kinetic
(2) The catalyst for the resolution must be readily available
resolution processes in general is the fact that the enantiomeric
in both enantiomeric forms. In the optimal case, the catalystwould be used in small quantities in the resolution and would
(4) (a) Katsuki, T. In ComprehensiVe Asymmetric Catalysis; Jacobsen, E. N.,
Pfaltz, A., Yamamoto, H., Eds.; Springer: New York, 1999; Chapter 18.1. (b) Rossiter, B. E. in Asymmetric Synthesis, Vol. 5; Morrison, J. D., Ed.;
(3) The nucleophile used for the ring opening should be
Academic Press: New York, 1985; Chapter 7. (c) Johnson, R. A.; Sharpless,
K. B. In Catalytic Asymmetric Synthesis; Ojima, I., Ed.; VCH: New York, 1993; Chapter 4.1. (d) Katsuki, T.; Martin, V. S. Org. React. 1996, 48, 1.
(4) The resolved epoxides must be obtained in good yield
(5) Reviews: (a) Jacobsen, E. N.; Wu, M. H. In ComprehensiVe Asymmetric
and very high enantiopurity and must be easily separated from
Catalysis; Jacobsen, E. N., Pfaltz, A., Yamamoto, H., Eds.; Springer: New York, 1999; Chapter 18.2. (b) Katsuki T. Coord Chem ReV. 1995, 140,
189. (c) Jacobsen, E. N. In ComprehensiVe Organometallic Chemistry II,
(5) Ideally, although not necessarily, the ring-opened byprod-
Vol. 12; Wilkinson, G., Stone, F. G. A., Abel, E. W., Hegedus, L. S., Eds.;Pergamon: New York, 1995; pp 1097-1135.
ucts should also be valuable chiral building blocks and be
(6) For a recent review: Frohn, M.; Shi, Y. Synthesis 2000, 1979.
obtainable in high enantiomeric excess.
(7) For asymmetric dihydroxylation routes, see: (a) Kolb, H. C.; Sharpless,
K. B. Tetrahedron 1992, 48, 10515. For asymmetric reduction methods,
To this end, we communicated recently the discovery that
see: (b) Corey, E. J.; Link, J. O. Tetrahedron Lett. 1991, 56, 442. (c) Corey, E. J.; Helal, C. J. Tetrahedron Lett. 1993, 34, 5227. (d) Ramachandran, P.
the (salen)Co complex 1 catalyzed the efficient hydrolytic kinetic
V.; Gong, B.; Brown, H. C. J. Org. Chem. 1995, 60, 41. (e) Kitamura, M.;
resolution (HKR) of a variety of terminal epoxides (Scheme
Tokunaga, M.; Noyori, R. J. Am. Chem. Soc. 1995, 117, 2931.
(8) For the most enantioselective methods developed to date involving synthetic
1).16-18 This new method appeared to hold considerable promise
catalysts: (a) Palucki, M.; Pospisil, P. J.; Zhang, W.; Jacobsen, E. N. J.
with regard to meeting all of the criteria outlined above. First,
Am. Chem. Soc. 1994, 116, 9333. (b) Collman, J. P.; Wang, Z.; Straumanis, A.; Quelquejeu, M.; Rose, E. J. Am. Chem. Soc. 1999, 121, 460. For methods involving biocatalysts, see: (c) Botes, A. L.; Weijers, C. A. G.
(14) Stereochemistry of Organic Compounds; Eliel, E. L., Wilen, S. H., Eds.;
M.; Botes, P. J.; van Dyk, M. S. Tetrahedron: Asymmetry 1999, 10, 3327,
and references therein. (d) Goswami, A.; Totleben, M. J.; Singh, A. K.;
(15) For an in depth discussion of practical considerations in kinetic resolution
Patel, R. N. Tetrahedron: Asymmetry 1999, 10, 3167, and references
reactions, see: Keith, J. M.; Larrow, J. F.; Jacobsen, E. N. AdV. Synth.,Catal. 2001, 343, 5-26.
(9) Gao, Y.; Klunder, J. M.; Hanson, R. M.; Masamune, H.; Ko, S. Y.;
(16) (a) Tokunaga, M.; Larrow, J. F.; Kakiuchi, F.; Jacobsen, E. N. Science
Sharpless, K. B. J. Am. Chem. Soc. 1987, 109, 5765. 1997, 277, 936. (b) Furrow, M. E.; Schaus, S. E.; Jacobsen, E. N. J. Org.
(10) Ladner, W. E.; Whitesides, G. M. J. Am. Chem. Soc. 1984, 106, 7250. Chem. 1998, 63, 6776.
(11) Hanson, R. M. Chem. ReV. 1991, 91, 437.
(17) For earlier studies involving (salen)metal-catalyzed reactions of epoxides
(12) Kasai, N.; Sakaguchi, K. Tetrahedron Lett. 1992, 33, 1211.
that served as a foundation for the discovery of the HKR, see: (a) Tekeichi,
(13) (a) Asymmetric ring opening of meso epoxides with TMSN3: Martı´nez,
T.; Arihara, M.; Ishimori, M.; Tsuruta, T. Tetrahedron 1980, 36, 3391. (b)
L. E.; Leighton, J. L.; Carsten, D. H.; Jacobsen, E. N. J. Am. Chem. Soc.
Maruyama, K.; Nakamura, T.; Nakamura, S.; Ogino, A.; Nishinaga, A. 1995, 117, 5897. (b) With carboxylic acids: Jacobsen, E. N., Kakiuchi, React. Kinet. Catal. Lett. 1991, 45, 165. (c) Larrow, J. F., Schaus, S. E.,
F., Konsler, R. G., Larrow, J. F., Tokunaga, M. Tetrahedron Lett. 1997,
Jacobsen, E. N. J. Am. Chem. Soc. 1996, 118, 7420. 38, 773. (c) With sulfides: Wu, M. H., Jacobsen, E. N. J. Org. Chem.
(18) The HKR is complementary to biocatalytic methods exploiting epoxide
1998, 63, 5252. (d) With TMSCN: Schaus, S. E.; Jacobsen, E. N. Org.
hydrolases. For a review, see: Archelas, A.; Furstoss, R. Trends Biotechnol.Lett. 2000, 2, 1001. 1998, 16, 108. 1308 J. AM. CHEM. SOC. 9 VOL. 124, NO. 7, 2002 HKR of Terminal Epoxides Catalyzed by (salen)CoIIIA R T I C L E S
racemic 1,2-epoxides are generally available directly from
commercial suppliers at low cost or are obtainable in one step from inexpensive R-olefins or aldehydes. In fact, certain racemic epoxides, such as propylene oxide, epichlorohydrin, styrene oxide, and butadiene monoepoxide, are commodity chemicals and are no more expensive than common organic solvents. Second, the ligands for catalyst 1 had previously been com- mercialized and manufactured on a ton scale in the context of (salen)Mn epoxidation catalysts.19 The cobalt analogues (R,R)-1 and (S,S)-1 proved equally accessible, and these are also now available in bulk.20 Third, water is perhaps the ideal reagent for effecting the resolution reaction: it is inexpensive and safe,
substrates. As a result of these efforts, we have discovered that
and the rate of the ring-opening reaction can be controlled
the HKR is an extraordinarily general reaction, allowing efficient
simply by modulating the rate of addition of water to the
kinetic resolution of virtually any type of terminal epoxide. Our
epoxide-catalyst mixture.21 Fourth, for those examples that
were described in the preliminary report, highly enantioenrichedepoxides were recovered from the HKR. Finally, the HKR
Results and Discussion
provided useful enantioenriched 1,2-diols, including many that
(I) Preparation of Catalyst and General Experimental
are otherwise not readily accessible using existing asymmetric
Considerations. Both enantiomers of the (salen)CoII complex 1 are available commercially on research or commercial scale,20
The HKR has seen rapid adoption as the method of choice
or they can be prepared from the commercially available ligands
for the preparation of a variety of terminal epoxides in
using Co(OAc)2 (see Experimental Section). The Co(II) complex
enantioenriched form, and a number of applications in target-
1 is catalytically inactive, however, and it must be subjected to
oriented synthesis have been reported already.23 In addition, the
one-electron oxidation to produce a (salen)CoIIIX complex
commercial manufacture of enantioenriched propylene oxide,
(X ) anionic ligand) prior to the HKR. This may be done
epichlorohydrin, and styrene oxide using HKR methodology has
conveniently by aerobic oxidation in the presence of a mild
been implemented, thereby reducing the cost of these useful
Brønsted acid. Water alone was found not to mediate the
chiral building blocks.20 We have sought to elucidate fully the
oxidation reaction, but a screen of additives revealed that acetic
synthetic potential of this reaction by establishing its substrate
acid was effective and that the corresponding Co(III) precatalyst
scope and outlining optimized procedures for the isolation of
1‚OAc is convenient for use in HKR reactions both in terms of
resolved epoxides and 1,2-diol products in high enantiomeric
excess. In that regard, we set as a common criterion for allsubstrates the isolation of resolved epoxide in >99% ee. We
1 + HOAc + (1/4)O
2 h 1‚OAc + (1/2)H2
also aimed to develop general protocols for the HKR that wouldallow the straightforward evaluation of previously unexamined
Two useful methods for the generation of complex 1‚OAc
have been developed. Method A involves isolation of 1‚OAc
(19) (a) Larrow, J. F.; Jacobsen, E. N.; Gao, Y.; Hong, Y.; Nie, X.; Zepp, C.
as a crude solid prior to the HKR. The Co(II) complex 1 is
M. J. Org. Chem. 1994, 59, 1939. (b) Larrow, J. F.; Jacobsen, E. N. Org. Synth. 1997, 75, 1.
dissolved in toluene to generate a ca. 1 M solution, and acetic
(20) For information, see: http://www.rhodiachirex.com.
acid (2 equiv) is added. The resulting solution is stirred open
(21) While it may be assumed that an “ideal” resolution would involve no added
reagentsi.e., an enantiomer undergoing selective isomerization or
to air at room temperature for 30 min, during which time the
polymerizationsthe rate of such transformation may be difficult to control
color of the mixture changes from orange to dark brown. All
because of the exothermicity (∆E > 30 kcal/mol) associated with epoxidering opening. This is a special concern with reactions carried out on a large
volatile materials are removed in vacuo, affording 1‚OAc as a
scale. The fact that the rate of nucleophile addition can be adjusted to control
brown solid residue that can be used without further purification.
reaction rate therefore has significant practical advantages.
(22) For the most effective catalyst developed thus far for the asymmetric
Method B involves in situ generation of 1‚OAc under HKR
dihydroxylation of terminal olefins, see: Becker, H.; Sharpless, K. B.
conditions by suspension of the Co(II) complex 1 in epoxide Angew. Chem., Int. Ed. Engl. 1996, 35, 448. For a general review of the AD reaction, see: Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B.
or epoxide/solvent and addition of HOAc under an aerobic
Chem. ReV. 1994, 94, 2483.
(23) (a) Schaus, S. E.; Brånalt, J. E.; Jacobsen, E. N. J. Org. Chem. 1998, 63,
4876. (b) Savle, P. S.; Lamoreaux, M. J.; Berry, J. F.; Gandour, R. D.
Catalyst obtained by both methods was examined for each
Tetrahedron: Asymmetry 1998, 9, 1843. (c) Gurjar, M. K.; Sadalapure, K.; Adhikari, S.; Sarma, B. V. N. B. S.; Talukdar, A.; Chorghade, M. S.
of the epoxides described in this study. For certain substrates
Heterocycles 1998, 48, 1471. (d) Gurjar, M. K.; Krishna, L. M.; Sarma, B.
such as 1-hexene oxide, catalyst prepared by either method leads
V. N. B. S.; Chorghade, M. S. Org. Proc. Res. DeV. 1998, 2, 422. (e) Cloninger, M. J.; Overman, L. E. J. Am. Chem. Soc. 1999, 121, 1092. (f)
to essentially identical results (Scheme 2). In these situations,
Rodrı´guez, A.; Nomen, M.; Spur, B. W.; Godfroid, J. J. Tetrahedron Lett.
in situ catalyst generation (method B) is preferable since the
1999, 40, 5161. (g) Hou, X.-L.; Li B.-F.; Dai, L.-X. Tetrahedron: Asymmetry 1999, 10, 2319. (h) Kamada, M.; Satoh, T.; Kakuchi, T.; Yokota, K. Tetrahedron: Asymmetry 1999, 10, 3667. (i) Yu, Q.; Wu, Y.; Xia, L.-
(24) The identity of the counterion can influence reactivity, enantioselectivity,
J.; Tang, M.-H.; Wu, Y.-L. Chem. Commun. 1999, 129. (j) Wyatt, P. B.;
and catalyst lifetime in the HKR. With the goal of defining general protocols
Blakskjær, P. Tetrahedron Lett. 1999, 40, 6481. (k) Liu, P.; Panek, J. S. J.
for HKR of the broadest range of substrates, we carried out all reactions
Am. Chem. Soc. 2000, 122, 1235. (l) Kno¨lker, H.-J.; Baum, E.; Reddy, K.
with catalyst 1‚OAc. However, other derivatives of 1 have been found to
R. Tetrahedron Lett. 2000, 41, 1171. (m) Wroblewski, A. E.; Halajewska-
display greater reactivity toward certain epoxides, and this is revealed most
Wosik, A. Tetrahedron: Asymmetry 2000, 11, 2053. (n) Liu, Z. Y.; Ji, J.
dramatically in the HKR of relatively unreactive substrates. For example,
X.; Li, B. G. J. Chem. Soc., Perkin Trans. 1 2000, 3519. (o) O’Neil, I. A.;
the HKR of methyl glycidate required use of 2 mol % 1‚OAc to provide
Cleator, E.; Southern, J. M.; Hone, N.; Tapolczay, D. J. Synlett 2000, 695.
recovered epoxide in 99% ee in 24 h (Table 4, entry 4). In contrast, use of
(p) Fu¨rstner, A.; Thiel, O. R.; Ackermann, L. Org. Lett. 2001, 3, 449. (q)
the p-nitrobenzoate complex 1‚O2CC6H4(NO2) at the 0.9 mol % level led
Chow, S.; Kitching, W. Chem. Commun. 2001, 1040. (r) Rodriguez, A.;
to >99% ee epoxide within the same time frame (C. P. Stevenson, work
Nomen, M.; Spur, B. W.; Godfroid, J. J.; Lee, T. H. Tetrahedron 2001,
in progress). The synthetic implications and mechanistic basis for these
effects are under investigation and will be described separately. J. AM. CHEM. SOC. 9 VOL. 124, NO. 7, 2002 1309 A R T I C L E S Table 1. Hydrolytic Kinetic Resolution (HKR) of Aliphatic Terminal
relative to the racemate. Similarly, the HKR of 1,2-epoxy-5-
hexene proved difficult to carry to completion under solvent-
free conditions, affording only 95% ee epoxide after 24 h.
However, using in situ generated catalyst (method B) and THF
as solvent (1:1, v/v, THF:H2O), epoxide could be recovered in
99.5% ee and 86% of the theoretical yield (entry 4). This
protocol was equally effective for the HKR epoxypropylbenzene
and vinylcyclohexane oxide, affording resolved epoxide in
g99% ee and 46% and 44% yield, respectively (entries 5,6).
Very hindered aliphatic epoxides such as tert-butylethylene
oxide proved to be particularly challenging substrates for the
a Reactions were carried out with 0.55 equiv of H2O relative to racemic
HKR, but efficient resolution was ultimately achieved through
epoxide. Water was added dropwise to a solution of catalyst and epoxide
careful optimization of reaction conditions. Under solvent-free
at 0 °C, and the reaction mixture was allowed to warm to room temperaturewith continuous stirring for the indicated time. b Catalyst loading based on
conditions employing up to 2 mol % catalyst generated by either
racemic epoxide. c In those cases where solvent was included (entries 3-7),
method A or B, no hydrolysis of tert-butylethylene oxide was
1:1 (v/v) ratio was used relative to epoxide. d Isolated yield of >99% ee
observed at room temperature over the course of several days.
epoxide based on racemic material (theoretical maximum ) 50%).
A systematic investigation of water-soluble organic solvents (e.g.
procedure avoids an extra solvent removal step. On the other
THF, i-PrOH, and 1,2-diols) revealed that the use of 1,2-
hand, catalyst prepared by method A was found to be more
hexanediol as solvent and 2 mol % 1‚OAc (generated by method
effective with less reactive substrates (vide infra) and was
A) was effective for inducing resolution of this epoxide to g99%
applicable to all substrates examined. Therefore, if HKR did
ee (entry 7). In general, such moderately lipophilic 1,2-diols
not afford epoxide in >99% ee with catalyst prepared by method
have proven quite effective as solvents for the most unreactive
B after optimization of solvent and catalyst loading, then catalyst
substrates in the HKR, presumably because of their ability to
effectively solubilize epoxide, water, and diol product.25
Aside from the method of generation of 1‚OAc, the only (b) Halogenated Epoxides. Three-carbon (C-3) epoxides
reaction parameters in the HKR that required optimization for
bearing halide substituents are highly versatile synthetic building
individual substrates were catalyst loading and choice of solvent.
blocks because each carbon is functionalized and a potential
With few exceptions, epoxide of >99% ee could be obtained
site of nucleophilic attack. Epichlorohydrin, in particular, is a
using 0.55 equiv of water relative to racemate. Relatively small
readily available C-3 unit that is widely employed in organic
epoxides with some degree of water solubility could be resolved
and polymer synthesis.26 However, this most interesting substrate
effectively without added solvent. However, the HKR of more
for the HKR initially proved problematic. It was found to
lipophilic substrates did benefit from inclusion of a water-
undergo gradual racemization under the reaction conditions,
miscible organic solvent such as tetrahydrofuran (THF), 2-pro-
thereby rendering it difficult to recover from HKR reactions in
panol, or 1,2-hexanediol. In general, one volume of solvent
highly enantioenriched form.16b The racemization pathway could
relative to racemic epoxides was sufficient to allow efficient
be suppressed, however, by carrying out the reaction at 0-4
HKR. Catalyst loadings of 0.5 mol % or lower relative to
°C in the presence of THF. Using 0.5 mol % 1‚OAc (generated
racemic epoxide were effective for many substrates, but epoxides
by method A), the epoxide could be recovered in g99% ee and
bearing sterically hindered or unsaturated substituents often
43% yield (Table 2, entry 1). In contrast, the HKR of
required more catalyst (up to 2 mol %) to attain complete
(()-epibromohydrin afforded the epoxide in 41% yield but only
resolution. Reactions were initiated at 0 °C and then allowed
43% ee under the same reaction conditions. In this case,
to warm to room temperature with continued stirring for 12-
bromide-catalyzed racemization could not be eliminated; how-
ever, it could be used to advantage in the dynamic HKR to
(II) HKR of Terminal Epoxides. (a) Aliphatic Epoxides.
produce diol (see section III). The HKR of (()-epifluorohydrin
As illustrated in Table 1, terminal aliphatic epoxides are
and (()-1,1,1-trifluoro-2,3-epoxypropane27 proceeded smoothly
outstanding substrates for the HKR, and for all substrates
under solvent-free conditions with no detectable racemization
examined the epoxide could be recovered in g99% ee and in
82-92% of the theoretical yield (41-46% yield based on
(c) Epoxides Bearing Ether and Carbonyl Functionality.
racemic epoxide). The HKR of propylene oxide proved to be a
The HKR was found to be applicable to a wide variety of ether
particularly efficient reaction, requiring only 0.2 mol % catalyst
containing epoxides (Table 3). Benzyl glycidyl ether, tert-
under solvent-free conditions and affording recovered epoxide
butyldimethylsilyl glycidyl ether, and phenyl glycidyl ether all
in high yield (entry 1). The HKR of 1-hexene oxide also
underwent resolution in excellent yield employing 0.5 mol %
proceeded well under solvent-free conditions (entry 2), but more
of the in situ generated catalyst 1‚OAc (method B; entries 1-3).
lipophilic epoxides with minimal solubility in water or the diol
1-Naphthyl glycidyl ether, a useful precursor to propranolol,28
product required the use of added solvents. For example, thediol product precipitates from the reaction mixture in the HKR
(25) There was no observable difference in the outcome of HKR reactions carried
of (()-1,2-epoxytetradecane under solvent-free conditions. This
out with racemic or enantiopure diols. This is consistent with the generalobservation of the absence of product inhibition in the HKR.
renders mixing extremely difficult in the late stages of the
(26) Huber, J. E. In Encyclopedia of Reagents for Organic Synthesis, Vol. 4;
resolution reaction and thereby leads to severely diminished
Paquette, L. A. Ed.; Wiley: New York, 1995; p 2326.
(27) For applications of this interesting building block, see: Katagiri, T.; Irie,
reaction rates. The HKR of this substrate was effected success-
M.; Uneyama, K. Org. Lett. 2000, 2, 2423, and references therein.
fully within 24 h using i-PrOH as the solvent at high initial
(28) (a) Klunder, J. M.; Ko, S. Y.; Sharpless, K. B. J. Org. Chem. 1986, 51,
3710. (b) For a report on the application of the HKR to aryl glycidyl ethers
concentration of epoxide (25 M), and employing 1 equiv of H2O
as a strategy for the synthesis of -blockers, see ref c. 1310 J. AM. CHEM. SOC. 9 VOL. 124, NO. 7, 2002 HKR of Terminal Epoxides Catalyzed by (salen)CoIIIA R T I C L E S Table 2. HKR of Halogenated Terminal Epoxidesa Table 5. HKR of Terminal, Conjugated Epoxidesa a Reactions were carried out with 0.55 equiv of H2O and 0.5 mol %
1‚OAc prepared from 1 by method A. Reactions were initiated at 0 °C and
then allowed to proceed at room temperature for 16-18 h. Reagent and
catalyst amounts based on racemic epoxide. b In those cases where solvent
was included (entries 1 and 2), 1:1 (v/v) ratio was used relative to epoxide. c Isolated yield based on racemic epoxide (theoretical maximum ) 50%). a Unless noted otherwise, reactions were carried out at 0 °C to room
temperature for 48 h, with 0.55 equiv of H2O relative to racemic epoxide
Table 3. HKR of Ether-Containing Terminal Epoxidesa
and THF (1:1 (v/v) with respect to epoxide) as solvent. b Catalyst loading based on racemic epoxide. In all cases, 1‚OAc was prepared by method A. c Isolated yield of >99% ee epoxide based on racemic material (theoretical
maximum ) 50%). d A 0.7 equiv amount of H2O used relative to racemic
epoxide; 72 h reaction time. e i-PrOH used as solvent (1:1 (v/v) relative to
The HKR of Boc protected 2,3-epoxy-1-aminopropane (entry
3) required relatively higher catalyst loading (2 mol % 1‚OAc)
and longer reaction time (48 h) to produce the epoxide in >99%
ee. The resolution of methyl glycidate (entry 4) also required
a Unless noted otherwise, reactions were carried out at 0 °C to room
2.0 mol % catalyst, with the enantioenriched epoxide recovered
temperature for 16-18 h, with 0.55 equiv of H2O relative to racemic epoxide
and THF (1:1 (v/v) with respect to epoxide) as solvent. b Catalyst loading
Epoxy ketones proved to be among the most problematic
based on racemic epoxide. c Isolated yield of >99% ee epoxide based onracemic epoxide (theoretical maximum ) 50%). d A 48 h reaction time.
substrates for the HKR. Under standard HKR conditions,
e A 0.6 equiv amount of H2O used relative to racemic epoxide. f The
substrates such as 3,4-epoxy-2-butanone (Table 4, entry 5)
substrate was D,L-butadiene diepoxide (Aldrich).
underwent only partial resolution, with the reduced, inactive
Table 4. HKR of Terminal Epoxy Esters, Ketones, and
Co(II) complex 1 precipitating from the reaction mixture within
a few hours. A stoichiometric Baeyer-Villiger-like pathway is
presumably responsible for reduction of the catalyst, although
no tractable byproducts could be identified. To maintain the
catalyst in the requisite Co(III) oxidation state, the HKR of
ketone-containing epoxides was carried out under an atmosphere
of O2. In the presence of 2 mol % AcOH and 0.7 equiv of H2O
and with 2 mol % 1‚OAc, 3,4-epoxy-2-butanone was recovered
in >99% ee and 40% isolated yield after 48 h.29 The HKR of
1,2-epoxy-3-pentanone (entry 6) was effected under similar
conditions to yield recovered epoxide in >99% ee and 41%
Unless noted otherwise, reactions were carried out at 0 °C to room
temperature for 16-18 h, with 0.55 equiv of H2O relative to racemic epoxide
and THF (1:1 (v/v) with respect to epoxide) as solvent. b Catalyst loading
(d) Aryl, Vinyl, and Alkynyl Epoxides. Styrene oxide
based on racemic epoxide. c Isolated yield of >99% ee epoxide based on
derivatives are among the most useful terminal epoxides from
racemic material (theoretical maximum ) 50%). d A 48 h reaction time. e A 24 h reaction time. f Reaction was carried out under an atmosphere of
a synthetic standpoint and are therefore particularly important
O2 (balloon pressure). g A 0.7 equiv amount of H2O used relative to racemic
candidates for the HKR reaction. In principle, HKR of these
substrates might be plagued by conflicting steric and electronic
required longer reaction times to attain high ee in the HKR,
factors influencing regioselectivity in the epoxide ring opening.
but was nevertheless resolved successfully (entry 4). The C-4
It was gratifying, therefore, to observe that resolution of
building block, (2-phenylmethoxymethyl)oxirane (entry 5) was
epoxides derived from various types of conjugated terminal
hydrolyzed efficiently using 0.5 mol % 1‚OAc to yield the
olefins (styrene, diene, and enyne derivatives) was possible with
enantioenriched epoxide in 42% yield. The HKR of com-
catalyst 1‚OAc using water-miscible solvents such as THF
mercially available (()-butadiene diepoxide was also effected
(Table 5). The HKR of (()-styrene oxide was effected using
successfully, requiring use of 1.0 mol % of 1‚OAc (prepared
0.8 mol % catalyst and 0.55 equiv of H2O, affording the
by method A) and 0.6 equiv of H2O to afford the recovered
recovered epoxide in 87% yield and >99% ee after 72 h (entry
diepoxide in >99% ee and 36% isolated yield. This interesting
1). Under similar conditions, both 3- and 4-chlorostyrene oxide
C-4 chiral building block has particular potential for elaboration
were obtained in >99% ee and 77 and 80% yield, respectively
to a variety of C2 symmetric diols.
(entries 2 and 3).30 Other 3-substituted styrene oxide derivatives
Epoxides containing carbonyl functionalities were also ex-
amined as substrates for the HKR (Table 4). The kinetic
(29) This reaction was applied as a key step in the total synthesis of the natural
product fostriecin: Chavez, D. E.; Jacobsen, E. N. Angew. Chem., Int. Ed.
resolutions of glycidyl butyrate and ethyl 3,4-epoxybutyrate
2001, 40, 3667.
(entries 1 and 2) were effected in a straightforward manner using
(30) For an account of the application of epoxidation/HKR protocols to the
preparation of 3-chlorostyrene oxide, see: Brandes, B. D.; Jacobsen, E. N.
0.5 mol % 1‚OAc (method B) in 46 and 44% yield, respectively. Tetrahedron: Asymmetry 1997, 8, 3927. J. AM. CHEM. SOC. 9 VOL. 124, NO. 7, 2002 1311 A R T I C L E S Figure 1. Plot of enantiomeric excess of recovered substrate as a function of conversion for representative k Figure 2. Plot of enantiomeric excess of product as a function of conversion
rel values as calculated using the equation
ln[(1 - c)(1 - eeSM)]/ln[(1 - c)(1 + eeSM)].34
rel values as calculated using the equation krel
ln[1 - c(1 + eeP)]/ln[1 - c(1 - eeP)].34
displayed similar reactivity in the HKR (entries 4 and 5). Incontrast, the resolution of 2-chlorostyrene oxide require in-
of substrates suggested the possibility that this reaction might
creased catalyst loading (1.5 mol % 1‚OAc) in order to attain
be applicable to product generation with synthetically useful
ee’s. We therefore undertook an investigation of the HKR of
The Eastman process for the low-cost aerobic epoxidation
terminal epoxides under conditions designed to allow isolation
of butadiene31 has rendered butadiene monoepoxide a practically
of 1,2-diol products with an optimal compromise of ee and yield.
useful building block for organic synthesis and an appealing
In general, satisfactory results were obtained in reactions
target for the HKR.32 The resolution of this substrate proceeded
employing 0.45 equiv of H2O relative to racemic epoxide.
in 72 h with 1.5 mol % 1‚OAc and 0.7 equiv of H
Reactions were usually complete within 12 h using the same
recovered epoxide in 99% ee and 36% yield (Table 5, entry 7).
catalyst loadings that had been identified in HKR’s in which
Alkynyl epoxides also appear to be good substrates for the HKR,
epoxide recovery was targeted (Table 6).
as evidenced by the successful resolution of the protected 1,2-
Outstanding results were obtained in the preparation of
epoxybutyne derivative in entry 8 using 0.8 mol % catalyst and
unhindered aliphatic 1,2-diols by the HKR procedure, with
products isolated in 99% ee and >40% yield. The observation
(III) Preparation of Enantioenriched 1,2-Diols via the
of such high product ee’s reflects selectivity factors in excess
HKR. As noted in the Introduction, one of the most attractive
of 300 for the HKR of these substrates (vide infra). Terminal,
features of kinetic resolution processes is the fact that ee of
unhindered olefins are among the poorest substrates for Os-
recovered starting substrate increases with conversion, and
catalyzed asymmetric dihydroxylation reactions,35 so the HKR
overresolution (i.e. reactions taken to >50% conversion) allows
methodology constitutes an especially interesting alternative for
production of very highly enantioenriched material even if the
the preparation of these important building blocks. While vinyl
resolution itself is only moderately selective. This is represented
cyclohexane oxide underwent HKR with similar success (entry
graphically in Figure 1, which depicts the familiar correlation
6), other, relatively hindered terminal aliphatic epoxides un-
between conversion and ee as a function of the selectivity factor
derwent hydrolysis with somewhat lower selectivity. Thus,
3-phenyl-1,2-propanediol was isolated in 95% ee and 40% yield,
kfast/kslow). As such, a kinetic resolution with a krel value
as low as 10 can provide recovered substrate in 95% ee and
as was the 1,2-diol derived from tert-butyloxirane (entries 5
34% yield. The situation with respect to product formation is
substantially different, however, since the ee of kinetic resolution
The HKR also provided practical access to a series of
product decreases with conversion. As reflected in the graph in
enantioenriched 1-halo-2,3-propane diol derivatives.16b Epichlo-
Figure 2, very high selectivity factors are required in order to
rohydrin underwent ring opening to afford 1-chloro-2,3-pro-
generate kinetic resolution products in high ee (e.g. >95%) and
panediol in 95% ee and 40% yield (Table 6, entry 8). However,
yields approaching 50%. For example, to obtain product with
direct distillation of the product from the reaction mixture
the same criteria outlined above (95% ee and 34% yield), a
resulted in deterioration of the ee of the product by as much as
selectivity factor of 63 would be required. For this reason, it
5%. An alternative isolation procedure was developed, wherein
comes as no surprise that the vast majority of kinetic resolutions
unreacted epoxide was removed by vacuum transfer, the reaction
involving synthetic catalysts has involved reactions targeting
residue was partitioned between hexanes/EtOAc (95:5) and H2O,
(33) Exceptions include: (a) Sharpless kinetic resolution: Gao, Y.; Hanson, R.
The fact that the HKR of terminal epoxides was observed to
M.; Klunder, J. M.; Ko, S. Y.; Masamune, H.; Sharpless, K. B. J. Am.
proceed with apparently very high selectivity for a broad range
Chem. Soc. 1987, 109, 5765. (b) Kinetic resolution of terminal epoxides with azide: Reference 17c. Kinetic resolution of terminal epoxides with phenols: Ready, J. M.; Jacobsen, E. N. J. Am. Chem. Soc. 1999, 121, 6086.
(31) Monnier, J. R. In 3rd World Congress on Oxidation Catalysis, 1997;
(c) Dynamic kinetic resolution processes (reviews): Noyori, R.; Tokunaga,
Grasselli, R. K., Oyama, S. T., Gaffney, A. M., Lyons, J. E., Eds.;
M.; Kitamura, M. Bull. Chem. Soc. Jpn. 1995, 68, 36. Strauss, U. T.; Felfer,
Elsevier: New York, 1997; pp 135-149.
U. Tetrahedron Asymmetry 1999, 10, 107. El Gihani, M. T.; Williams, J.
(32) An effective dynamic kinetic resolution of butadiene monoepoxide with
M. J. Curr. Opin. Chem. Biol. 1999, 3, 11.
inorganic carbonates has been developed: Trost, B. M.; McEachern, E. J.
(34) Kagan, H. B.; Fiaud, J. C. in Topics in Stereochemistry, Vol. 14; Eliel, E. J. Am. Chem. Soc. 1999, 121, 8649. While this method does not allow
L., Wilen, S. H., Eds., Wiley: New York, 1987; pp 249-330.
isolation of recovered epoxide in the enantioenriched form, it provides an
(35) See: Reference 22. Vanhessche, K. P. M.; Sharpless, K. B. Chem. Eur. J.
attractive approach to the corresponding diol. 1997, 3, 517. 1312 J. AM. CHEM. SOC. 9 VOL. 124, NO. 7, 2002 HKR of Terminal Epoxides Catalyzed by (salen)CoIIIA R T I C L E S Table 6. Synthesis of Enantioenriched 1,2-Diols via the HKR of Scheme 3. Double Resolution Route to 1-Chloro-2,3-propanediol Scheme 4. Preparation of Enantioenriched Glycidol from the
Epoxides Bearing Ether and Carbonyl Functionality
in high ee (Table 2, entry 2). However, with 0.45 equiv of H2O,
diol was obtained in 96% ee. These results suggested that
epibromohydrin might be undergoing racemization with adven-
titious bromide ion under the conditions of the HKR, raising
the possibility that dynamic kinetic resolution may be possible.36
Indeed, this turned out to be the case: the dynamic kinetic
resolution was accomplished using 2 mol % (R,R)-1‚OAc
(method A), 1.5 equiv of H2O in THF (5.0 M) at 0 °C to yield
(R)-1-bromo-2,3-propanediol in 96% ee and 90% yield. Both
1-bromo- and 1-chloro-2,3-propanediol are useful intermediates
for the preparation of glycidol and its derivatives (Scheme 4).37
As summarized in Table 6, the HKR of nearly all terminal
epoxides examined proceeded effectively under standard condi-
tions to afford 1,2-diol products in 94-99% ee and in good
yield. Among the exceptions, the HKR of (()-glycidyl butyrate
yielded the corresponding 1,2-diol in only 43% ee upon isolation
Unless noted otherwise, reactions were carried out for 12-14 h with
0.45 equiv of H2O relative to racemic epoxide. Water was added dropwise
(entry 18). Given the fact that under similar conditions epoxide
to a solution of catalyst and epoxide at 0 °C, and the reaction mixture was
can be recovered in >99% ee (Table 4, entry 1), it appears likely
allowed to warm to room temperature and stir for the indicated time. b
that the diol is undergoing racemization during the HKR by a
Catalyst loading based on racemic epoxide. c In those cases where solvent
was included, 1:1 (v/v) ratio was used relative to racemic epoxide. d Isolated
transesterification pathway. The HKR of the Boc protected
yield of diol based on racemic material (theoretical maximum ) 45%).e A
3-amino-1,2-epoxypropane (entry 20) afforded the diol in 36%
1.5 equiv amount of H2O used relative to racemic epoxide. The theoretical
yield and only 78% ee. This particular substrate appears to be
yield in this dynamic kinetic resolution reaction is 100%. f A 0.4 equivamount of H2O used relative to racemic epoxide. g The substrate was D,L-
one of the poorest for this resolution process of those examined.
butadiene diepoxide. h A 5 h reaction time. (IV) Determination of krel Values. Because the ee’s of
starting material and product change as a function of conversion,
and the mixture filtered to break up the emulsion. The organic
it is often most useful to characterize kinetic resolution reactions
layer was separated and extracted further with H2O, and the
not in terms of the ee obtained but rather in terms of the relative
combined aqueous extracts were concentrated to yield pure
reaction rates of the two enantiomeric substrates (k
1-chloro-2,3-propanediol with no deterioration of ee. The HKR
kslow). Assuming a first-order kinetic dependence on these
of epifluorohydrin and trifluoropropylene oxide proceeded
substrates,38 the relationship between the conversion, c, of the
effectively to yield the corresponding diols in 97% ee (38%
reaction and the ee of the of the unreacted substrate and of the
yield) and >99% ee (42% yield), respectively.
product formed is straightforward and depicted graphically in
In cases where 1,2-diol of very high enantiomeric excess is
Figures 1 and 2, respectively. The practical matter of determin-
required, it is a straightforward matter to effect a “double
ing accurate krel values is clearly dependent on accurate
resolution” wherein highly enantioenriched epoxide is obtainedby methods outlined in part II and then subjected to a second
(36) For a review, see: Noyori, R.; Tokunaga, M.; Kitamura, M. Bull. Chem.Soc. Jpn. 1995, 68, 36.
HKR with the opposite enantiomer of catalyst. This approach
(37) The HKR of glycidol itself provided resolved epoxide in low (<20%) yield
is illustrated in Scheme 3 in the context of the preparation of
as a result of the participation of undesired oligomerization pathways. It isinteresting to note, however, that epoxide ring opening has not been
1-chloro-2,3-propanediol of >99% ee.
observed in any cases with the 1,2-diol products of the HKR.
(38) Different ee vs conversion curves are obtained in kinetic resolutions with
The HKR of epibromohydrin proved particularly interesting.
kinetic dependencies on substrate other than 1. See: Luukas, T. O.; Girard,
As noted in section IIb, this was the only substrate examined
C.; Fenwick, D. R.; Kagan, H. B. J. Am. Chem. Soc. 1999, 121, 9299. Johnson, D. W., Jr.; Singleton, D. A. J. Am. Chem. Soc. 1999, 121, 1,
that failed to undergo resolution to provide recovered epoxide
J. AM. CHEM. SOC. 9 VOL. 124, NO. 7, 2002 1313 A R T I C L E S
measurement of both ee and conversion, and for highly selective
Table 7. Determination of kRel Values in the HKR of Terminal
ments can translate to large errors in the calculation of the
It became clear with experimentation that highly reproducible
measurements of ee ((0.1%) could be obtained by capillary
GC analysis of epoxide or diol products, but substantially greater
variability was observed in measurements of conversion.
Analysis of Figures 1 and 2 reveals that, in highly selective
kinetic resolutions, lowest sensitivity to errors in the measure-
ment in conversion is attained with evaluation of product at
low conversions. For consistency, we carried out all measure-
ments by effecting HKR reactions using 0.20 equiv of H
relative to racemic epoxide. The conversions were then deter-
mined by measuring the isolated yield of pure 1,2-diol. As a
Epoxides Bearing Ether and Carbonyl Functionality
result, the values for conversion c listed in Table 7 represent
lower limits, as do the values calculated for krel.
carrying out reproducibility studies on vinylcyclohexane oxide
(entry 6), one of the best substrates for the HKR. It was found
that the yield varied (2% and the ee varied by (0.1% in seven
resolutions carried out under identical conditions. These varia-
tions result in calculated krel values ranging from 490 to 840.
Thus, while the relative magnitudes of the values in Table 7
provide useful guidelines for evaluation of the HKR, the absolute
magnitudes for the best substrates are certainly lacking precision.
Nevertheless, the data in Table 7 highlight one of the principal
features of the HKR: all practical issues notwithstanding, this
reaction is one of the most selective asymmetric catalytic
reactions discovered to date. The observation of k
excess of 100 for a broad range of substrates is remarkable,
cases point to a nearly perfect chiral recognition mechanism
a Unless noted otherwise, reactions were carried out with 0.2 equiv of
H2O relative to racemic epoxide. Water was added dropwise to a solution
(V) Catalyst Recycling. The possibility of recycling a catalyst
of catalyst 1‚OAc (prepared by method B) and epoxide at 0 °C, and the reaction mixture was allowed to warm to room temperature and stir for 12
has obvious practical appeal, particularly in cases where the
h. Catalyst loadings and solvents were identical to those used in Table 6
catalyst is precious due to cost or limited availability. Catalyst
for the same substrates. b Isolated yield of 1,2-diol. c Calculated using the
1 is prepared in bulk from low-cost components, and as a result
ln[1 - c(1 + eeP)]/ln[1 - c(1 - eeP)]. d The substrate was
D,L-butadiene diepoxide. e Because the diol product from this reaction was
it is quite inexpensive relative to most chiral catalysts. On the
susceptible to racemization, determination of krel was made by evaluating
other hand, the HKR employs reactants (racemic epoxide, water,
unreacted epoxide using standard preparative conditions and applying the
minimal if any solvent) that impact the cost of the overall
ln[(1 - c)(1 - eeSM)]/ln[(1 - c)(1 + eeSM)].
process to an almost negligible extent in many cases, and as a
Scheme 5. Catalyst Recycling in the HKR of Propylene Oxide
result the catalyst is a significant contributor to the materialcosts. Accordingly, efforts were directed toward identifyingpractical methods for effecting catalyst recovery and recycling.
The HKR reaction of propylene oxide presents an especially
straightforward scenario with respect to catalyst recoverybecause both the epoxide and the diol are relatively volatileand can be removed by distillation. The solid residue remainingin the reaction vessel after product separation was found to havethe characteristic red-brick color of the reduced (salen)CoII
A more ambitious test of the recyclability of the HKR catalyst
complex 1. Reoxidation to 1‚OAc with air and AcOH (method
was undertaken wherein each subsequent cycle was carried out
B) led to catalyst with undiminished levels of reactivity and
with a different substrate (Table 8). Starting with 400 µmol of
(R,R)-1 (242 mg), six HKR reactions were carried out sequen- tially, with epoxide isolated by vacuum transfer, and diol isolated
(39) For a lucid analysis of kinetic resolutions with either enantiopure or
enantioimpure catalysts and of the obstacles to obtaining accurate measure-
either by vacuum distillation or by trituration.41 Recovered
ments of krel, see: Blackmond, D. G. J. Am. Chem. Soc. 2001, 123, 545.
catalyst was reactivated with air and HOAc as a common
(40) In the description of this result in the initial report on the HKR (ref 16a,
Scheme 2), the absolute stereochemistries of the epoxide and diol productswere accidentally reversed. The stereochemistries indicated here in Scheme
(41) Complete experimental details of the recycling experiments are provided
in the Supporting Information Experimental Section. 1314 J. AM. CHEM. SOC. 9 VOL. 124, NO. 7, 2002 HKR of Terminal Epoxides Catalyzed by (salen)CoIIIA R T I C L E S Table 8. HKR with Catalyst Recyclinga Experimental Section
Complete experimental procedures for all substrates are provided
[(R,R)-N,N′-Bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanedi- aminato(2-)]cobalt(II) ((R,R)-1). A solution of cobalt(II) acetate
tetrahydrate (5.98 g, 24.0 mmol) in MeOH (80 mL was added to a
solution of ligand [(R,R)-N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-
cyclohexanediamine]46 (10.9 g, 20.0 mmol) in CH2Cl2 (80 mL) via
cannula under an atmosphere of N2 with careful exclusion of air. A
brick-red solid began to precipitate before addition was complete. The
a Reactions were carried out with 0.55 equiv of H
sides of the reaction flask were rinsed with MeOH (20 mL), and the
epoxide. Water was added dropwise to a solution of catalyst and epoxide
mixture was allowed to stir for 15 min at room temperature and then
at 0 °C, and the reaction mixture was allowed to warm to room temperature
30 min at 0 °C. Precipitated solids were isolated by vacuum filtration
with continuous stirring for 16-72 h. b Catalyst loading based on racemic
and rinsed with cold (0 °C) MeOH (2 × 75 mL). The red solid was
epoxide. The entire batch of catalyst (400 µmol) was used in eachexperiment, with the amount of other reagents adjusted accordingly. c In
collected and dried in vacuo to yield [(R,R)-N,N′-bis(3,5-di-tert-
those cases where solvent was included, 1:1 (v/v) ratio was used relative
butylsalicylidene)-1,2-cyclohexanediaminato(2-)]cobalt(II) ((R,R)-1)
to epoxide. d Isolated yield of >99% ee epoxide based on racemic material
Representative Procedures for the HKR of Terminal Epoxides.
intermediate step. Again, no loss of catalytic activity or
(a) Method A. (S)-Propylene Oxide. A 100 mL flask equipped with a stir bar was charged with (S,S)-1 (242 mg, 400 µmol, 0.002 equiv).
enantioselectivity was observed. At the end of the sixth cycle,
The catalyst was dissolved in 5 mL of PhMe and treated with AcOH
the catalyst was isolated by filtration to yield (R,R)-1 in 88%
(240 µL, 4.2 mmol). The solution was allowed to stir at room
recovery (212 mg, 351 µmol).
temperature open to air for 30 min over which time the color changed
Conclusions and Outlook
from orange-red to a dark brown. The solution was concentrated in
The extraordinarily high levels of selectivity observed in the
vacuo to leave a crude brown solid. The resulting catalyst residue was
HKR raise interesting questions about the mechanism of
dissolved in propylene oxide (14.0 mL, 11.6 g, 200 mmol) at roomtemperature, the reaction flask was cooled to 0 °C, and H
catalysis. While a full investigation will be the topic of an
110 mmol, 0.55 equiv) was added dropwise over 5 min. The reaction
upcoming, separate paper, it is clear from preliminary kinetic
was allowed to warm to room temperature and stir 14 h at which time
studies that the reaction follows a second-order dependence on
(S)-propylene oxide (5.35 g, 92.1 mmol, 46%) was isolated by
catalyst concentration.16a This is consistent with observations
distillation from the reaction mixture at atmospheric pressure and 36
made in (salen)Cr-catalyzed reactions of epoxides with azide42
°C. Propylene diol was removed by vacuum distillation (65 °C, 0.25
and suggests a cooperative, bimetallic mechanism for the
Torr). The catalyst was recovered by suspension in MeOH and
selectivity-determining epoxide ring-opening event.43 This
collection by vacuum filtration. The ee of the propylene oxide was
insight has led to the design and development of multimeric
determined to be 99.7% by chiral GC analysis of the 1-azido-2-
(salen)Co catalysts with dramatically enhanced reactivitysand
trimethylsiloxypropane derivative obtained by opening the epoxide with
in some cases improved enantioselectivity
3 (Cyclodex-B, 55 °C, isothermal, tR(minor) ) 12.29 min,
opening reactions.44 These new generation catalysts are interest-
(b) Method B. (R)-1,2-Epoxy-5-hexene. A 100 mL flask equipped
ing both on a fundamental and a practical level with regard to
with a stir bar was charged with (R,R)-1 (302 mg, 500 µmol, 0.005
the future elucidation and development of the HKR and related
equiv). The catalyst was treated with (()-1,2-epoxy-5-hexene (11.3
reactions. On the other hand, the monomeric catalyst 1 displays
mL, 9.81 g, 100 mmol), AcOH (120 µL, 2.1 mmol, 0.02 equiv), and
broad effectiveness for the selective hydrolysis of racemic,
1 mL of THF. The reaction flask was cooled to 0 °C, and H2O (1.0
terminal epoxides, and it holds special appeal due to its
mL, 55 mmol, 0.55 equiv) was added in one portion. The reaction was
simplicity and ready availability at low cost. Thus, it is likely
allowed to warm to room temperature and stir 16 h at which time the
that catalyst 1 will remain the system of choice for HKR
volatile materials were isolated by vacuum transfer at 0.25 Torr into a
reactions, on a laboratory scale in particular.
cooled (-78 °C) receiving flask. The recovered epoxide was filtered
The HKR provides a straightforward method for the prepara-
through a silica plug to remove residual water, and the THF was
tion of a wide assortment of terminal epoxides in highly
removed by rotary evaporation to yield (R)-1,2-epoxy-5-hexene (4.23g, 43.1 mmol). The diol was distilled under reduced pressure (56 °C,
enantioenriched form.45 Given that in many cases there exist
0.25 Torr). The catalyst was recovered by suspension in MeOH and
no practical alternatives for accessing the valuable chiral
vacuum filtration. The ee of the recovered epoxide was determined to
building blocks, it is hoped that the HKR will have a beneficial
be 99.5% by chiral GC analysis of the 1-azido-2-trimethylsiloxy-5-
and enabling effect on the field of organic synthesis.
hexene derivative obtained by opening the epoxide with TMSN3(Cyclodex-B, 70 °C, isothermal, tR(minor) ) 38.00 min, tR(major) )
(42) Hansen, K. B.; Leighton, J. L.; Jacobsen, E. N. J. Am. Chem. Soc. 1996,
(43) For other examples of catalytic asymmetric ring opening of epoxides
Acknowledgment. This work was supported by the NIH
involving cooperative effects, see: (a) Iida, T.; Yamamoto, N.; Matsunaga, S.; Woo, H.-G.; Shibasaki, M. Angew. Chem., Int. Ed. Engl. 1998, 37,
(Grant GM-43214). We thank the Ford Foundation for a
2223. (b) McCleland, B. W.; Nugent, W. A.; Finn, M. G. J. Org. Chem.1998, 63, 6656.
(44) (a) Polymer-bound catalysts: Annis, D. A.; Jacobsen, E. N. J. Am. Chem.Supporting Information Available: Complete experimental Soc. 1999, 121, 4147. (b) Dendrimeric catalysts: Breinbauer, R.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2000, 39, 3604. (c) Oligomeric catalysts:
procedures and characterization data (PDF). This material is
Ready, J. M.; Jacobsen, E. N. J. Am. Chem. Soc. 2001, 123, 2687.
available free of charge via the Internet at http://pubs.acs.org.
(45) Thus far, efforts to extend the HKR to other classes of racemic epoxides
have proven unsuccessful, although our efforts continue in this direction.
For kinetic resolution of 2,2-disubstituted epoxides with TMSN3 catalyzed by the chromium analogue of 1, see: Lebel, H.; Jacobsen, E. N. Tetrahedron
(46) Larrow, J. F.; Jacobsen, E. N. Org. Synth. 1997, 75, 1. Also available Lett. 1999, 40, 7303. J. AM. CHEM. SOC. 9 VOL. 124, NO. 7, 2002 1315
COMMITTENTE OGGETTO DEL SERVIZIO INTERVENTO PROGETTUALE Progetto per la costruzione del secondo tratto della strada di P.R.G. che collega laVia F. Crispi alla piazza G. Bonaccorso con annesso parcheggio nel Comune di AciBonaccorsi Progetto per la realizzazione di una tomba a cielo scoperto per complessivi numero 4posti sita nel cimitero comunale di Acireale lato OvestProgetto Per la r
PLANNED PARENTHOOD (PPSTSCHS) Corporate Office: 3601 Fannin, Houston, TX 77004 [713-522-6240] DISCHARGE MEDICATIONS Doxycycline 100mg. Start today. Take one blue capsule twice a day for 7 days. Take 15 to 20 minutes after eating a solid food. Doxycycline can cause nausea and vomiting if taken on an empty stomach. Metronidazole 500mg. Take all four white pills together tonight af
 A R T I C L E S
A R T I C L E S HKR of Terminal Epoxides Catalyzed by (salen)CoIII
A R T I C L E S
HKR of Terminal Epoxides Catalyzed by (salen)CoIII
A R T I C L E S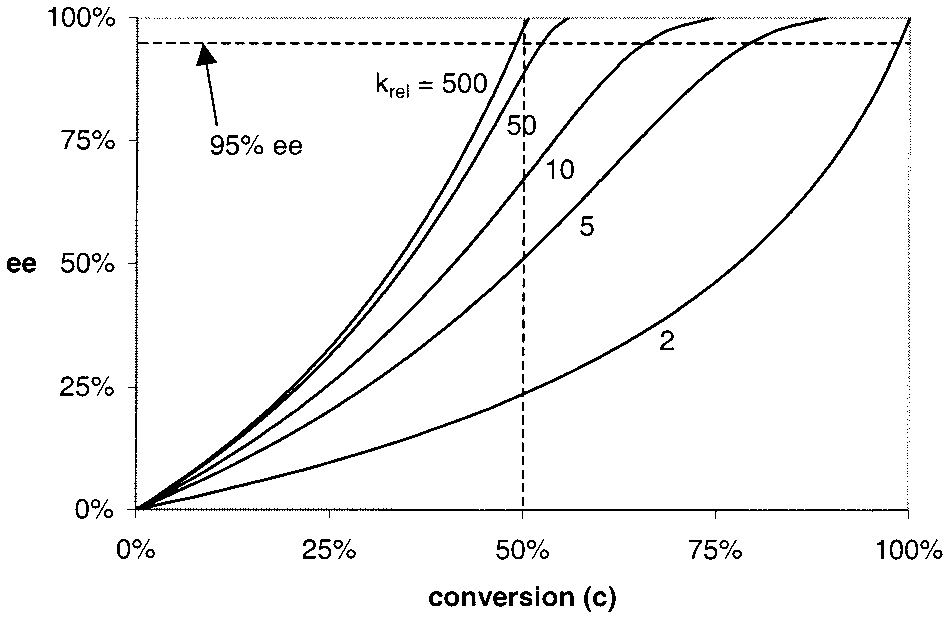
 A R T I C L E S
A R T I C L E S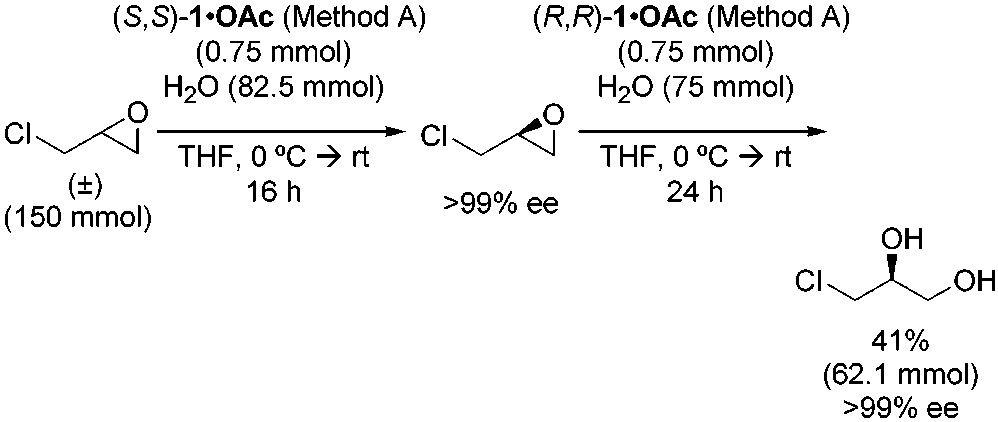
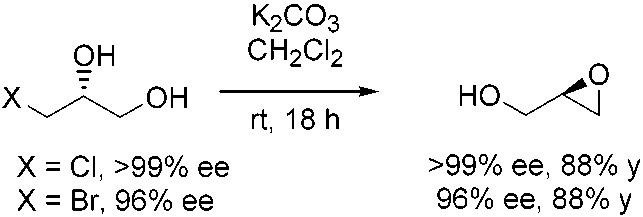 HKR of Terminal Epoxides Catalyzed by (salen)CoIII
A R T I C L E S
HKR of Terminal Epoxides Catalyzed by (salen)CoIII
A R T I C L E S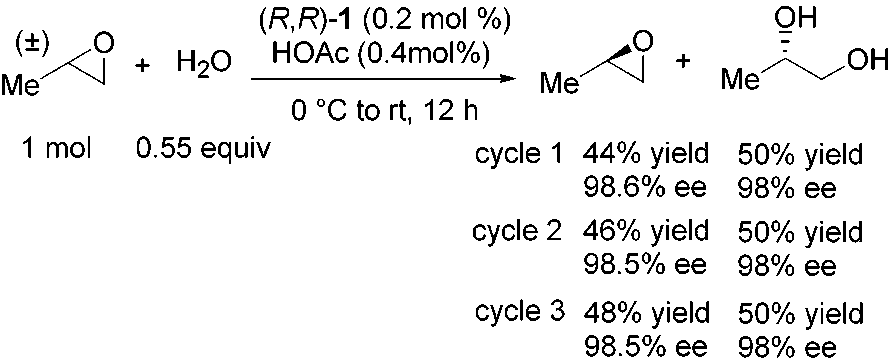 A R T I C L E S
A R T I C L E S